rMATS v4.0.2 (turbo)
Xing Lab, Children's Hospital of Philadelphia
- Which version to use
- Installation
- Test rMATS v4.0.2 (turbo) on PC3E & GS689 dataset:
- Using rMATS v4.0.2 (turbo)
- Output
- Alternative Splicing Events
Which version to use:
rMATS v4.0.2 (turbo) was built with two different settings of Python interpreter. In order to know which version you should use, you need to check what Unicode type your Python is built with. Open python console and type in:
>>> import sys
>>> print sys.maxunicode
1114111
This output indicates that your python is built with --enable-unicode=ucs4, and you should use rMATS-turbo-xxx-UCS4.
>>> print sys.maxunicode
1114111
>>> import sys
>>> print sys.maxunicode
65535
This output indicates that your python is built with --enable-unicode=ucs2, and you should use rMATS-turbo-xxx-UCS2.
>>> print sys.maxunicode
65535
Installation:
|
# For CentOS 6:
pip install numpy
yum install lapack-devel blas-devel
yum install gsl-devel.x86_64
yum install gcc-gfortran
# For Ubuntu 14:
pip install numpy
sudo apt-get install libblas-dev liblapack-dev
sudo apt-get install libgsl0ldbl
sudo apt-get install gfortran
# For Mac OS X Yosemite 10.10.5 (Using Homebrew for package management):
brew install gcc@5
brew install gsl
pip install numpy
pip install numpy
yum install lapack-devel blas-devel
yum install gsl-devel.x86_64
yum install gcc-gfortran
# For Ubuntu 14:
pip install numpy
sudo apt-get install libblas-dev liblapack-dev
sudo apt-get install libgsl0ldbl
sudo apt-get install gfortran
# For Mac OS X Yosemite 10.10.5 (Using Homebrew for package management):
brew install gcc@5
brew install gsl
pip install numpy
|
|
|
|
tar -xzf rMATS.4.0.2.tgz
cd rMATS.4.0.2/
... # move/copy/download data to this folder.
tar -xzf gtf.tgz
tar -xzf testData.tgz
cd rMATS.4.0.2/
... # move/copy/download data to this folder.
tar -xzf gtf.tgz
tar -xzf testData.tgz
Test rMATS v4.0.2 (turbo) on PC3E & GS689 dataset:
Run rmats.py as below to test rMATS runs properly.
cd rMATS.4.0.2/
python rMATS-turbo-xxx-UCSx/rmats.py --b1 b1.txt --b2 b2.txt --gtf gtf/Homo_sapiens.Ensembl.GRCh37.75.gtf --od bam_test -t paired --readLength 50 --cstat 0.0001 --libType fr-unstranded
Output can be found in the bamTest directories. The test run output should look like the rMATS output description.python rMATS-turbo-xxx-UCSx/rmats.py --b1 b1.txt --b2 b2.txt --gtf gtf/Homo_sapiens.Ensembl.GRCh37.75.gtf --od bam_test -t paired --readLength 50 --cstat 0.0001 --libType fr-unstranded
Using rMATS v4.0.2 (turbo):
The following is a detailed description of the options used with rMATS v4.0.2 (turbo).
Usage:
Optional:
Examples:
Usage:
Running with fastq
Running with bam
Required Parameters:
python rmats.py --s1 s1.txt --s2 s2.txt --gtf gtfFile --bi STARindexFolder -od outDir -t readType -readLength readLength [options]*
Running with bam
python rmats.py --b1 b1.txt --b2 b2.txt --gtf gtfFile --od outDir -t readType --nthread nthread --readLength readLength --tstat tstat [options]*
| --s1 s1.txt | A text file contains FASTQ file(s) for the sample_1.(Only if using fastq) |
| --s2 s2.txt | A text file contains FASTQ file(s) for the sample_2.(Only if using fastq) |
| --b1 b1.txt | A text file records mapping results for the sample_1 in bam format. (Only if using bam) |
| --b2 b2.txt | A text file records mapping results for the sample_2 in bam format. (Only if using bam) |
| -t readType | Type of read used in the analysis. readType is either 'paired' or 'single'. 'paired' is for paired-end data and 'single' is for single-end data |
| --readLength <int> | The length of each read |
| --gtf gtfFile | An annotation of genes and transcripts in GTF format |
| --bi STARIndexFolder | The folder name of the STAR binary indexes (i.e., the name of the folder that contains SA file). For example, use ~/STARindex/hg19 for hg19. (Only if using fastq) |
| --od outDir | The output directory |
| --tophatAnchor <int> | The "anchor length" or "overhang length" used in the aligner. At least “anchor length” NT must be mapped to each end of a given junction. The default is 6. (This parameter applies only if using fastq) |
| --nthread <float> | The number of thread. The optimal number of thread should be equal to the number of CPU core. |
| --cstat <float> | The cutoff splicing difference. The cutoff used in the null hypothesis test for differential splicing. The default is 0.0001 for 0.01% difference. Valid: 0 ≤ cutoff < 1 |
| --tstat <float> | The number of thread for statistical model. |
| --statoff | Turn statistics part off. |
| -libType libraryType | Library type. Default is unstranded (fr-unstranded). Use fr-firststrand or fr-secondstrand for strand-specific data. |
Example using fastq.
$cat s1.txt:
231ESRP.25K.rep-1.R1.fastq:231ESRP.25K.rep-1.R2.fastq,231ESRP.25K.rep-2.R1.fastq:231ESRP.25K.rep-2.R2.fastq
$cat s2.txt:
231EV.25K.rep-1.R1.fastq:231EV.25K.rep-1.R2.fastq,231EV.25K.rep-2.R1.fastq:231EV.25K.rep-2.R2.fastq
$
231ESRP.25K.rep-1.R1.fastq:231ESRP.25K.rep-1.R2.fastq,231ESRP.25K.rep-2.R1.fastq:231ESRP.25K.rep-2.R2.fastq
$cat s2.txt:
231EV.25K.rep-1.R1.fastq:231EV.25K.rep-1.R2.fastq,231EV.25K.rep-2.R1.fastq:231EV.25K.rep-2.R2.fastq
$
python rMATS-turbo-xxx-UCSx/rmats.py --s1 s1.txt --s2 s2.txt --gtf gtf/Homo_sapiens.Ensembl.GRCh37.72.gtf --bi ~/STARindex/hg19 --od out_test -t paired --nthread 6 --readLength 50 --tophatAnchor 8 --cstat 0.0001 --tstat 6
Example using bam.
$cat b1.txt:
231ESRP.25K.rep-1.bam,231ESRP.25K.rep-2.bam
$cat b2.txt:
231EV.25K.rep-1.bam,231EV.25K.rep-2.bam
$
231ESRP.25K.rep-1.bam,231ESRP.25K.rep-2.bam
$cat b2.txt:
231EV.25K.rep-1.bam,231EV.25K.rep-2.bam
$
python rMATS-turbo-xxx-UCSx/rmats.py --b1 b1.txt --b2 b2.txt -gtf gtf/Homo_sapiens.Ensembl.GRCh37.75.gtf -od bam_test -t paired --readLength 50 --cstat 0.0001 --libType fr-unstranded
Output:
All output files are in --od which contains rMATS output of AS events, all possible alternative splicing (AS) events derived from GTF and RNA
|
|
{kind=link}
|
|
|
|
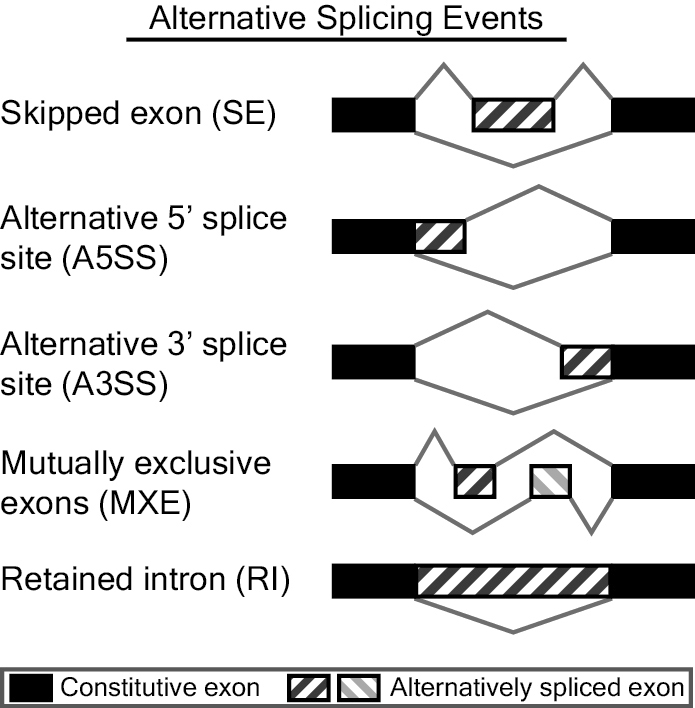
Alternative Splicing Events
rMATS analyzes skipped exon (SE), alternative 5' splice site (A5SS), alternative 3' splice site (A3SS), mutually exclusive exons (MXE), and retained intron (RI) events. Possible alternative splicing
events are identified from the RNA-Seq data and annotation of transcripts in GTF format. The following is a list of provided GTF files:
|
Alternatively, you can download your own transcript annotation in GTF format. However, the first column (chromosome/contig name) in the GTF must match the sequence names in your STARindex.