Install MATS:
- Add the Python directory to the $PATH environment variable
|
- Add the bowtie and tophat directories to the $PATH environment variable
|
- Add the samtools directory to the $PATH environment variable
|
- Obtain bowtie index for genome by either of the following two ways
- Build own bowtie index using bowtie-build from genome fasta sequence
|
|
- Untar MATS and bowtie indexes. For example, assuming you use pre-build bowtie indexes, unpack
MATS.3.0.0.beta.tgz in your home directory and unpack bowtieIndexes.tgz in your
bowtieIndexes directory:
|
cd
tar -xvzf MATS.3.0.0.beta.tgzcd ~/bowtieIndexestar -xvzf bowtieIndexes.tgz
Test MATS:
Run testRun.sh as below to test MATS runs properly.
cd ~/MATS.3.0.0.beta
./testRun.sh ~/bowtieIndexes/hg19
This two outputs can be found in the fastqTest and bamTest directories. The test run output should look like the
MATS output description.
Trim Fastq (Optional):
To trim the poor quality 3' end of reads, use the trimFastq.py script found in the bin directory.
python trimFastq.py input.fastq trimmed.fastq desired_length
cd ~/MATS.3.0.0.beta/
python bin/trimFastq.py testData/231EV.25K.rep-1.R1.fastq testData/trimmed.fastq 32
The above command trims 231EV.25K.rep-1.R1.fastq to 32 bp long by removing sequence from the 3' end of the reads and then saves it to trimmed.fastq.
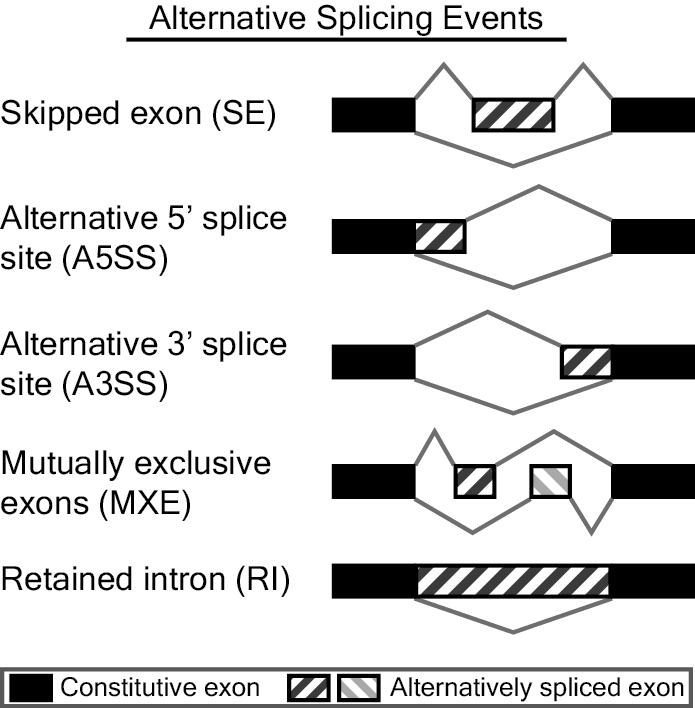
Alternative Splicing Events
MATS analyzes skipped exon (SE), alternative 5' splice site (A5SS), alternative 3' splice site (A3SS), mutually exclusive exons (MXE), and retained intron (RI) events. Possible alternative splicing
events are identified from the RNA-Seq data and annotation of transcripts in GTF format. The following is a list of provided GTF files found in the gtf directory:
- Human, Homo sapiens (Ensembl or UCSC Known Genes)
|
- Mouse, Mus musculus (Ensembl or UCSC Known Genes)
|
- Drosophila, Drosophila melanogaster (FlyBase)
|
- C. Elegans, Caenorhabditis elegans (RefSeq)
|
- Zebrafish, Danio rerio (RefSeq)
|
Alternatively, you can download your own transcript annotation in GTF format. However, the first column (chromosome/contig name) in the GTF
must match the sequence names in your bowtie
index. Use
bowtie-inspect (found in the bowtie directory) to display sequence names for the bowtie index.
bowtie-inspect --names your_bowtie_index
Only use a GTF in which the chromosome/contig name (first column) matches with the above command output.
Using MATS:
The following is a detailed description of the options used with MATS.
Usage:
Running with
fastq
python RNASeq-MATS.py -s1 rep1_1[:rep1_2][,rep2_1[:rep2_2]]* -s2 rep1_1[:rep1_2][,rep2_1[:rep2_2]]* -gtf gtfFile -bi bowtieIndexBase -o outDir -t readType -len readLength [options]*
Running with
bam
python RNASeq-MATS.py -b1 s1_rep1.bam[,s1_rep2.bam]* -b2 s2.rep1.bam[,s2.rep2.bam]* -gtf gtfFile -o outDir -t readType -len readLength [options]*
| -s1 rep1_1[:rep1_2][,rep2_1[:rep2_2]]* | FASTQ file(s) for the sample_1. For the paired-end data, two files must be colon separated and replicates must be in a comma separated list (Only if using fastq) |
| -s2 rep1_1[:rep1_2][,rep2_1[:rep2_2]]* | FASTQ file(s) for the sample_2. For the paired-end data, two files must be colon separated and replicates must be in a comma separated list (Only if using fastq) |
| -b1 s1_rep1.bam[,s1_rep2.bam] | Mapping results for the sample_1 in bam format. Replicates must be in a comma separated list (Only if using bam) |
| -b2 s2.rep1.bam[,s2.rep2.bam] | Mapping results for the sample_2 in bam format. Replicates must be in a comma separated list (Only if using bam) |
| -t readType | Type of read used in the analysis. readType is either 'paired' or 'single'. 'paired' is for paired-end data and 'single' is for single-end data |
| -len <int> | The length of each read |
| -gtf gtfFile | An annotation of genes and transcripts in GTF format |
| -bi bowtieIndexBase | The basename of the bowtie indexes (ebwt files). The base name does not include the first period. For example, use hg19 for hg19.1.ebwt. (Only if using fastq) |
| -o outDir | The output directory |
Optional:
| -a <int> | The "anchor length" used in TopHat. At least “anchor length” NT must be mapped to each end of a given junction. The default is 8 |
| -r1 <float>[,<float>]* | The insert sizes of sample_1 data. This applies only for the paired-end data. Replicates are separated by comma. The default is 15 for each replicate |
| -r2 <float>[,<float>]* | The insert sizes of sample_2 data. This applies only for the paired-end data. Replicates are separated by comma. The default is 15 for each replicate |
| -sd1 <float>[,<float>]* | The standard deviations for the r1. Replicates are separated by comma. The default is 70 for each replicate |
| -sd2 <float>[,<float>]* | The standard deviations for the r2. Replicates are separated by comma. The default is 70 for each replicate |
| -c <float> | The cutoff splicing difference. The cutoff used in the null hypothesis test for differential splicing. The default is 0.05 for 5% difference. Valid: 0 ≤ cutoff < 1 |
| -analysis analysisType | Type of analysis to perform. analysisType is either 'P' or 'U'. 'P' is for paired analysis and 'U' is for unpaired analysis. The default is 'U' |
| -expressionChange <float> | Filters out AS events with whose gene expression levels differs more than the given cutoff fold change between the two samples. Valid: fold change > 1.0. The default is 10.0 |
Examples:
Example using
fastq, performing
unpaired analysis
python RNASeq-MATS.py -s1 testData/231ESRP.25K.rep-1.R1.fastq:testData/231ESRP.25K.rep-1.R2.fastq,testData/231ESRP.25K.rep-2.R1.fastq:testData/231ESRP.25K.rep-2.R2.fastq -s2 testData/231EV.25K.rep-1.R1.fastq:testData/231EV.25K.rep-1.R2.fastq,testData/231EV.25K.rep-2.R1.fastq:testData/231EV.25K.rep-2.R2.fastq -gtf gtf/Homo_sapiens.Ensembl.GRCh37.65.gtf -bi ~/bowtieIndexes/hg19 -o out_test -t paired -len 50 -a 8 -r1 72,75 -sd1 40,35 -r2 70,65 -sd2 48,45 -c 0.1 -analysis P -expressionChange 20.0
Example using
bam, performing
paired analysis
python RNASeq-MATS.py -b1 testData/231ESRP.25K.rep-1.bam,testData/231ESRP.25K.rep-2.bam -b2 testData/231EV.25K.rep-1.bam,testData/231EV.25K.rep-2.bam -gtf gtf/Homo_sapiens.Ensembl.GRCh37.65.gtf -o bam_test -t paired -len 50 -a 8 -c 0.1 -analysis U -expressionChange 20.0
Output:
All output files are in outputFolder
- MATS_output: A folder that contains MATS output of AS events. Each output file is sorted by P-values in acending order.
- AS_Event.MATS.JunctionCountOnly.txt evaluates splicing with only reads that span splicing junctions
- IJC_SAMPLE_1: inclusion junction counts for SAMPLE_1, replicates are separated by comma
|
- SJC_SAMPLE_1: skipping junction counts for SAMPLE_1, replicates are separated by comma
|
- IJC_SAMPLE_2: inclusion junction counts for SAMPLE_2, replicates are separated by comma
|
- SJC_SAMPLE_2: skipping junction counts for SAMPLE_2, replicates are separated by comma
|
|
- AS_Event.MATS.ReadsOnTargetAndJunctionCounts.txt evaluates splicing with reads that span splicing junctions and reads on target (striped regions on home page figure)
- IC_SAMPLE_1: inclusion counts for SAMPLE_1, replicates are separated by comma
|
- SC_SAMPLE_1: skipping counts for SAMPLE_1, replicates are separated by comma
|
- IC_SAMPLE_2: inclusion counts for SAMPLE_2, replicates are separated by comma
|
- SC_SAMPLE_2: skipping counts for SAMPLE_2, replicates are separated by comma
|
|
- Important columns contained in both types of output files
- IncFormLen: length of inclusion form, used for normalization
|
- SkipFormLen: length of skipping form, used for normalization
|
- IncLevel1: inclusion level for SAMPLE_1 replicates (comma separated) calculated from normalized counts
|
- IncLevel2: inclusion level for SAMPLE_2 replicates (comma separated) calculated from normalized counts
|
- IncLevelDifference: average(IncLevel1) - average(IncLevel2)
|
|
|
- summary.txt: A file that contains summary of statistically significant AS events and the identity of each replicate
|
- ASEvents: A folder that contains all possible alternative splicing (AS) events derived from GTF and RNA
|
- SAMPLE_1/REP_N: A folder that contains mapping results of sample_1, replicate N
- accepted_hits.bam is the original tophat output containing both multi-mapped and uniquely mappable reads.
|
- unique.S1.sam contains uniquely mappable reads only. MATS uses uniquely mappable reads.
|
|
- SAMPLE_2/REP_N: A folder that contains mapping results of sample_2, replicate N
- accepted_hits.bam is the original tophat output containing both multi-mapped and uniquely mappable reads.
|
- unique.S2.sam contains uniquely mappable reads only. MATS uses uniquely mappable reads.
|
|
- commands.txt: A list of key commands executed
|
- log.RNASeq-MATS: Log file for running MATS pipeline
|
{kind=link}